Summary: This tutorial explains how to submit count files (output of Oasis' sRNA Detection module) into the Differential Expression (DE) module. The DE Analysis module is the second analysis module of Oasis and it calculates differential expression, makes target predictions, and provides functional analyses of sRNAs. In brief, the module returns quality metrics of your samples with respect to their different conditions, detailed differential expression results, known and predicted miRNA targets, and gives the user the opportunity to analyze various gene ontology (GO) and pathway enrichments. Apart from the regular DE analysis of two groups, this module supports complex DE analyses, ranging from multi-group comparisons (see section Submit DE analysis) to the incorporation of covariate information (see Part 2 below). Guidelines on how to interpret the results of the DE module can be found in Oasis' DE Output Tutorial.
The DE Analysis module requires count files as input, where those files are output by the sRNA Detection module. The count files, being associated with samples from different conditions (e.g. different time- points , biological experiments , etc.), are input as either "control" or "treatment" to execute the DE analysis. Be aware that although it is safer to submit count files from a single sRNA Detection analysis, it is not required (as long as the submitted samples are from the same organism). Also, novel miRNAs might not be considered in the DE Analysis if they are not present in both sets of count files.
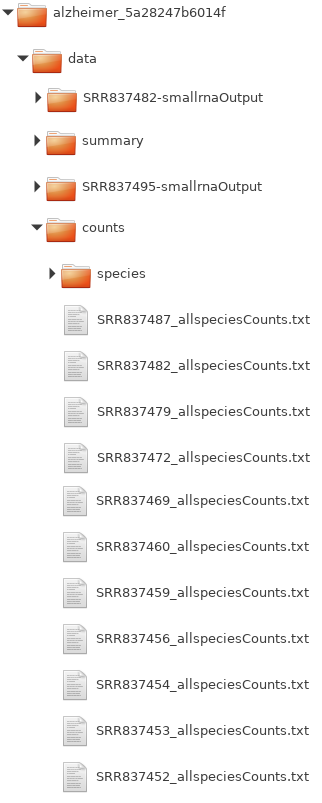
Within the directory of the output from the sRNA detection module, you will find the data folder containing various subfolders pertaining to the global and individual results of the different samples (Fig. ) (see the sRNA Output Tutorial on how to obtain it). The count files can be found within the subdirectory "counts" as text files containing sRNA IDs and read counts for those sRNAs.
Those count files should be uploaded as control and treatment samples to run the DE analysis.
This part will show you how to submit jobs to Oasis’ DE Analysis module. It will cover the submission of jobs for two or more conditions. More sophisticated differential expression analyses (covariate information) will be covered later in this tutorial.
All the steps required to submit a job are performed on the web interface of the DE Analysis module (Fig. , red border).
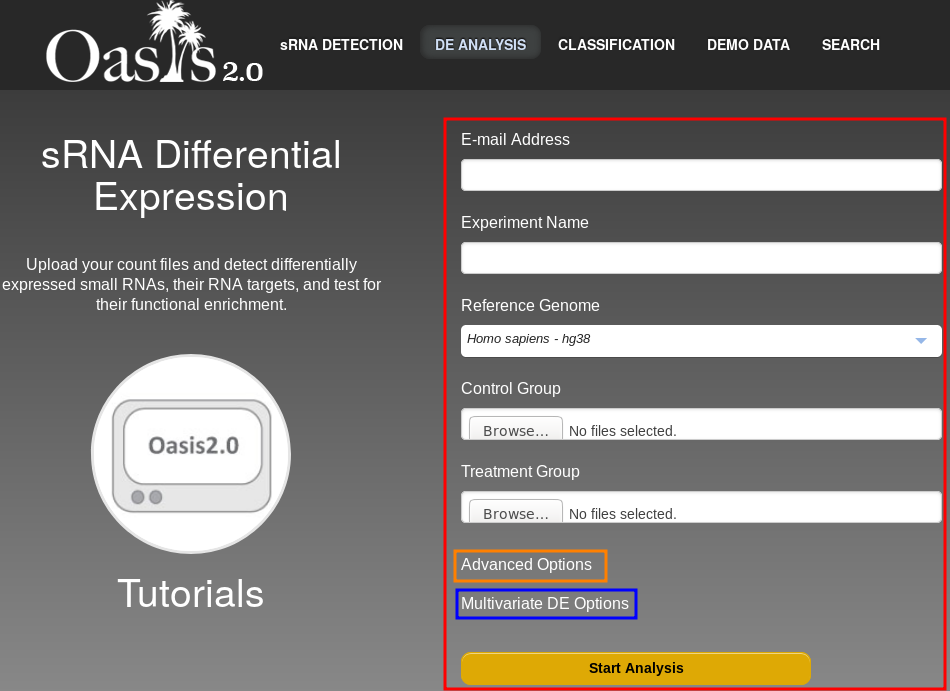
The individual fields are as follows:
This covers the basic (mandatory) parameters and files that you have to provide.
There are further options that might be of interest to you. If you click on Advanced options (marked in orange in figure ) you can select the Base mean cutoff value which sets the threshold for the minimum number of times (counts) a sRNA has to be in your dataset for it to be considered for the differential expression analysis.
In some experiments, additional factors may have had an influence on your experiments, such as age or gender of patients. This information can be accounted for and "corrected" by means of creating a covariate information file and entering a design formula.
As an example, assume you work with human samples that fall into two classes: Alzheimer Disease and healthy samples. You would like to know which sRNAs change with AD, but you also know that the data might vary depending on the age and gender of the individuals. As such, the DE analysis module can calculate the differential expression of sRNAs based on the disease conditions, while correcting for the age and gender factors.
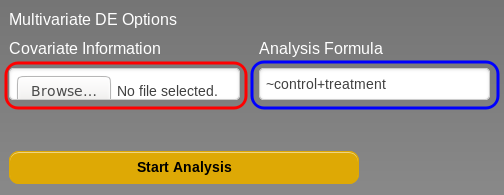
You can open the advanced options by clicking on the "Multivariate DE Options" link in the main page (Fig . , blue box). This will open two additional fields that will allow you to upload a file containing sample covariate information (Fig. , red box) and an analysis formula (Fig. , blue box).
The file with the covariate information should have tab-separated-values (TSV), with the columns indicating sample names and conditions and covariates being tested and corrected for, respectively. Table shows an example of a covariate information file with a disease phenotype, gender and age, based on our demo Alzheimer’s data (Leidinger et al., 2013). In general, the first column should contain the file names as they appear in the count files that you uploaded before. The other columns should contain the covariate information and conditions to be tested.
| ID | DiseasePheno | Gender | Age |
|---|---|---|---|
| Sample_SRR837437_allspeciesCounts.txt | AD | F | 77 |
| Sample_SRR837438_allspeciesCounts.txt | AD | M | 74 |
| Sample_SRR837439_allspeciesCounts.txt | AD | M | 68 |
| Sample_SRR837440_allspeciesCounts.txt | AD | F | 75 |
| Sample_SRR837441_allspeciesCounts.txt | AD | M | 75 |
| Sample_SRR837442_allspeciesCounts.txt | AD | M | 76 |
| Sample_SRR837443_allspeciesCounts.txt | AD | M | 79 |
| Sample_SRR837444_allspeciesCounts.txt | AD | F | 75 |
| Sample_SRR837445_allspeciesCounts.txt | AD | M | 77 |
| Sample_SRR837446_allspeciesCounts.txt | AD | F | 75 |
| Sample_SRR837447_allspeciesCounts.txt | AD | M | 76 |
The design formula (Fig. , blue box) specifies how Oasis will analyse your data. In table , in in order to correct for the gender and age covariates while testing for the disease condition, the formula should look as such: ~Gender+Age+DiseasePheno Note that the variable of interest should be the right-most one in the design formula and that variable names need to be written exactly as they appear in the covariate information file (i.e. case-sensitive). You should also be aware that if you do not include Table 1, a formula in the box, the analysis will create a design formula based on the order of the columns in the covariate information file. For the design formula applied by default would be ~DiseasePheno+Age+Gender (i.e. testing for gender, while correcting for disease phenotype and age).
For more information on the covariate analysis and the DE analysis in general, please refer to the DESeq2 documentation (Love, Huber, & Anders, 2014).