Summary: This tutorial explains how to submit the output of small RNA (sRNA) deep sequencing experiments (sRNA-seq) into the sRNA Detection module of Oasis. sRNA detection is the first analysis module of Oasis, and it examines sample qualities, as well as quantifies known and novel sRNAs for each submitted sample. This module supplies valuable information on the quality of samples, and returns count files that can be submitted to Oasis' DE Analysis module (differential sRNA expression analysis) or the Classification module (biomarker detection). This tutorial aims at getting the user familiar with submitting FastQ files generated from sRNA-seq to Oasis. In order to understand how to interpret the results of this module, please refer to the sRNA Output Tutorial videos and PDFs.
Oasis works with FASTQ files that you should have obtained from your sequencing run. Each experimental sample should be a single FASTQ file. You can either upload one such FASTQ file individually or you can bundle them all as a zip file. However, a better solution is often times to use the Oasis compressor to reduce the size of your files. This will make uploading your files a lot faster. You can download the Oasis compressor here. Then you can proceed to the Oasis compressor section of this document go learn how to use it.
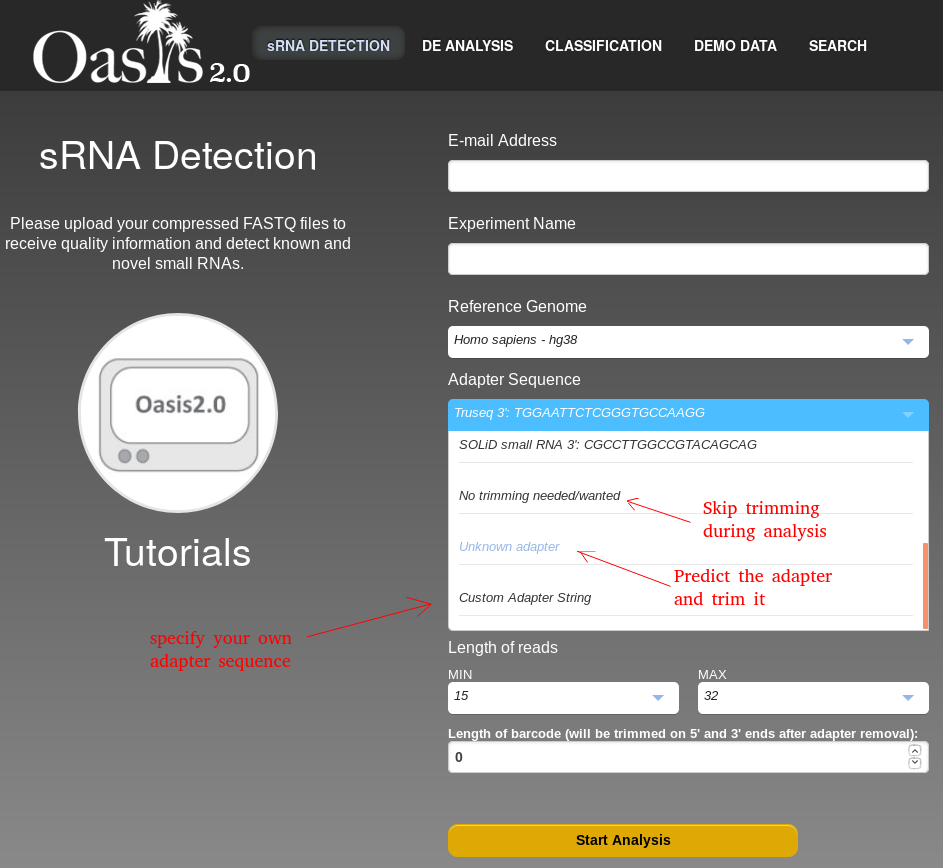
When you want to start your analysis job with Oasis, you need to fill out a form that provides the details about how your job should be run. First of all, you need to specify an e-mail address. Status updates and the results will be sent to this e-mail address so that you will later know where you can download the results of the Oasis analysis. The second form field is the Experiment's Name. Here you should insert a descriptive name so that you will later be able to recognize this analysis job among other analysis jobs (assuming that you will analyse several independent experiments). Next you have to select a reference genome which will be used for aligning your sequencing reads and determining what the reads represent. You also have to specify the 3'-sequencing adapter (if you don't know which adapter was used, ask whoever sequenced the samples). Alternatively, if you do not know the adapter, Oasis can try to determine the adapter for you. Simply select Unknown adapter. If you know which adapter was used and you want to specify a specific adapter sequence, select Custom Adapter String and enter the sequence. Otherwise, if you already trimmed the sequencing adapter, you can select no trimming needed/wanted. These options are also shown in Figure . Finally you have to specify the input files. All input files have to be either FASTQ files or (a collection of) zipped FASTQ files. The allowed file extensions are .fastq, .fastq.gz. Alternatively you can also upload the files that were compressed by the Oasis compressor.
There are also some advanced options that you might be interested in. First of all, you can specify the fraction of mismatches that are allowed when the reads are mapped to the genome. These are expressed as percentages of the total read length, as reads vary in length after trimming (default: 5% of the read length, i.e. reads of lengths 15- 19 are allowed 0 mismatches and reads of lengths 20-32 are allowed 1 mismatch). Next, you can specify the (minimum and maxium) length of reads that are allowed for your analysis. Too short reads are likely to be mapped to the wrong locations and too long reads probably contain sequencing artifacts. Finally, if your sequencing experiment used a barcode, you can specify the length of the barcode. The barcode will be trimmed from both sides of the reads.
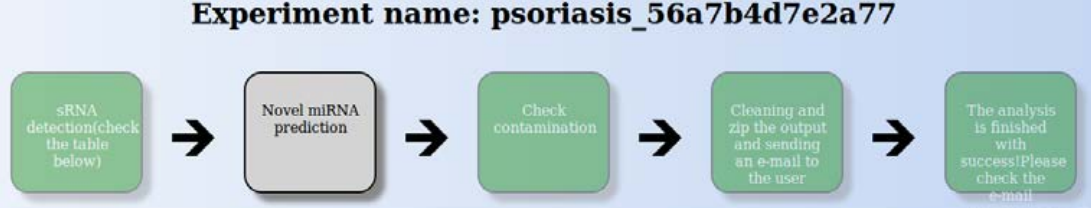
Once the analysis starts, you will get an e-mail indicating the analysis started, along with information about the analysis parameters. In addition, the e-mail will contain a link allowing you to follow the progress of the analysis. This includes a diagram showing the step the analysis is at (sRNA detection, novel miRNA prediction, contamination check and cleaning, zipping and sending the output to the user), with the current step marked with grey background (Fig. ).
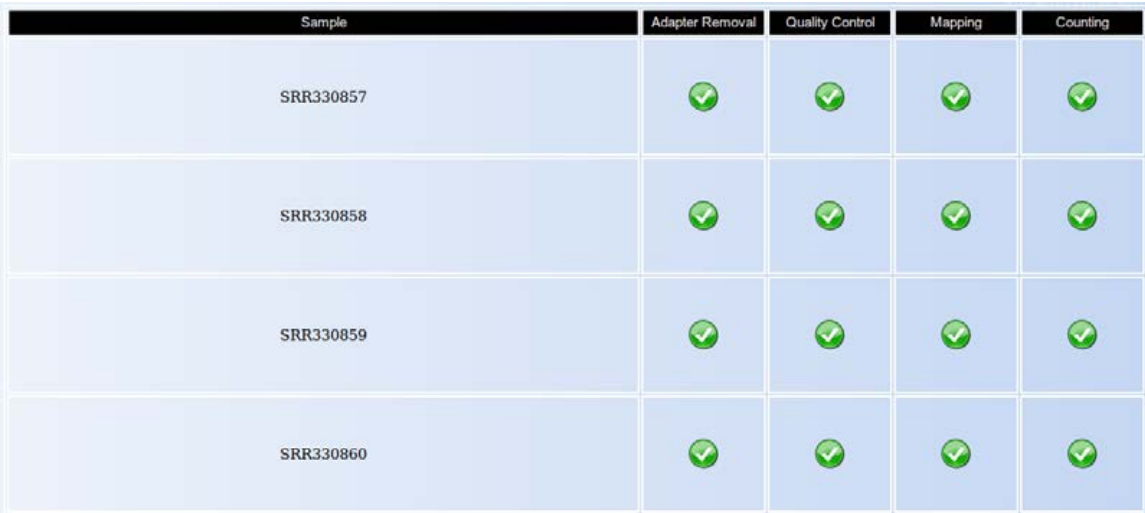
This step involves removing the 3' adapter, the quality control, the mapping to the reference genome and the counting of reads in each sRNA for each sample. While the analysis runs for a particular sample, an hourglass icon is shown, and when the analysis is done, a checkmark is shown.
This step involves detecting new miRNAs by predicting their structures and genomic location. For further elaboration on this technique, please refer to the sRNA Output Tutorial videos and PDFs.
All reads that were unaligned to the genome are re-aligned to viral genomes in order to determine the extent of viral contamination within the miRNA data.
The data is put into a directory hierarchy and an HTML file is constructed for it. The directory is then zipped, and an e-mail is sent to the user with a link to download the zip file. To see how to analyse the results, please refer to the sRNA Output Tutorial videos and PDFs.
After downloading and saving OasisCompressor.jar on your computer, double-click to launch its Graphical User Interface to submit your samples
Select your FastQ files by pressing on the ‘Input file(s)' button. For this, you can select a folder containing all files or several files separately. Note that OasisCompressor only accepts files of type fastq or fastq.gz (e.g. sample_name.fastq.gz).
Create an empty directory with the name of the experiment, then press the Output Directory button and select the empty directory to store the compressed FastQ.
Lastly, you can select the number of parallel processes OasisCompressor will launch by selecting the Core 1 dropdown menu. OasisCompressor allows you to select all cores available on your computer, although we recommend using NO MORE THAN 80% of all available cores. This will allow your computer to run other tasks while OasisCompressor is running.
Click Start Compression. Depending on the size of your files and the speed of your computer, OasisCompressor will take 20-60 minutes to compress your samples. Once it finishes, you can upload the compressed .zip file to Oasis' sRNA Detection module.